Functional studies
eQTL
eQTL analysis was one of the first approaches that we used to understand the underlying functional mechanisms of regions associated with
myeloma risk. The difficulty in performing an eQTL in MM is the influence on expression of global hidden co-variants both experimental and biological. In order to correct for such
factors, Probabilistic estimation of expression residuals (PEER) Bayesian Framework - was utilised
with known covariates (batch, gender, population eigenvector days in post, cell count) and hidden factors. PEER derived covariates were calculated and applied as
covariates in testing by linear regression with packages such as MatrixQTL. or
FastQTL. Subsequently, six independent eQTLs were identified within the
23 known risk regions. A recent follow-up
transcriptome-wide association study (TWAS) found 13 independent regions the included 108 genes. Identification of
eQTLs/MeQTLs/pQTLs will be important for inferring/imputing phenotypes from large sequencing projects and machine learning approaches will become common,
e.g. tensorflow implementation of FastQTL , PrediXcan or Fusion
and Sequential regulatory activity prediction. I have been familiarising myself with these approaches with both sequencing and
array data.
The top right figure shows a strong myeloma eQTL from 180 UK and 660 German matched normal SNP data with myeloma plasma cell expression. Bottom right is a regional plot together with
a conditional analysis. The top left is the correlation matrix of PEER derived co-factors together with known factors. Bottom left is a table of genome-wide significant
myeloma GWAS associations with significant eQTLs in bold.

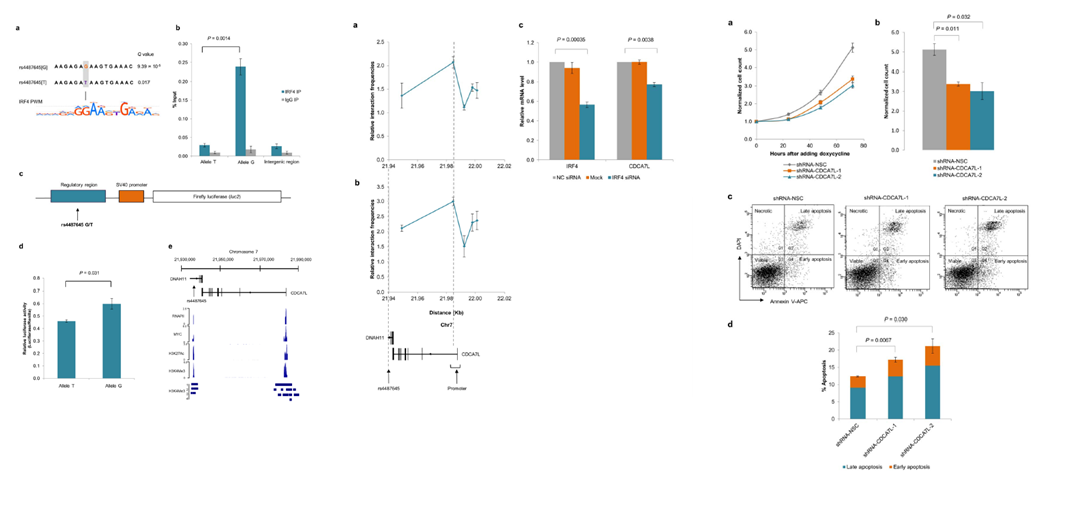
CDCA7L-IRF4-MYC
CDCA7L was the first Myeloma significantly association eQTL linked with a genome-wide risk allele.
Fine mapping, conditional testing and regulatory annotation with DNase hypersensitivity and ChIP-seq tracks confirmed the strongest association was the most likely
candidate for the causal region. We next performed luciferase reporter assays to determine the effect of rs4487645 on enhancer activity in the MM cell line KMS11.
Transfection of constructs containing the risk G-allele showed a significant enhancement of normalized luminescence compared with the T-allele (figure on left). 3C-qPCR showed that
rs4487645 physically interacts with the CDCA7L promoter in GM11992 and KMS11. siRNA knockdown of IRF4 gave a subsequent lower expression in CDCA7L (middle figures).
Increased CDCA7L expression was associated with increased cell proliferation and poorer patient survival. CDCA7L knockdown induces cell apoptosis and suppresses cellular proliferation
as assessed by FACS using Annexin (figures on the right). Thus, we demonstrate that differential IRF4 binding rs4487645 influences the expression of c-Myc-interacting CDCA7L.
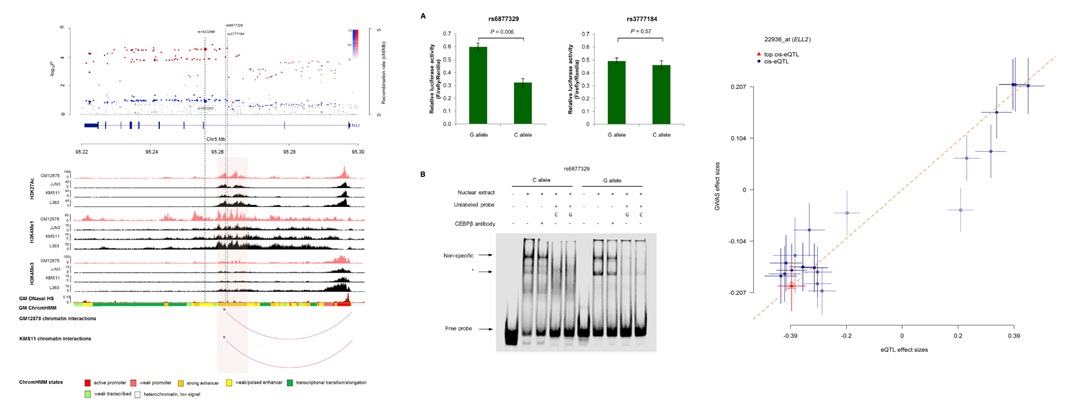
ELL2
Fine mapping of ELL2 association region excluded the present of a rare disease causing coding variant. Stratified eQTL and regulatory annotation with DNase hypersentivity and ChIP-seq tracks indicated a chromatin-looping interaction
with the ELL2 promoter and the strongest association (left figure below). We performed luciferase reporter assays in KMS11 . Transfection with constructs containing the
rs6877329-C risk allele displayed significantly lower normalized luminescence compared to non-risk G-allele construct (two-tailed t test p = 0.006). We performed a
electrophoretic mobility shift assay (EMSA) with CEBPB antibody (middle figure below). Here we demonstrated that rs6877329 resides within an enhancer that physically interacts
with the ELL2 promoter. SThe rs6877329-C risk allele reduces enhancer activity and is associated with reduced ELL2 expression in myeloma patients. Figure on the right shows effect sizes
of SNPs (used for the HEIDI test) from GWAS meta-analysis plotted against those for SNPs from the respective eQTL study in a summary data-based Mendelian randomization
(SMR) analysis.
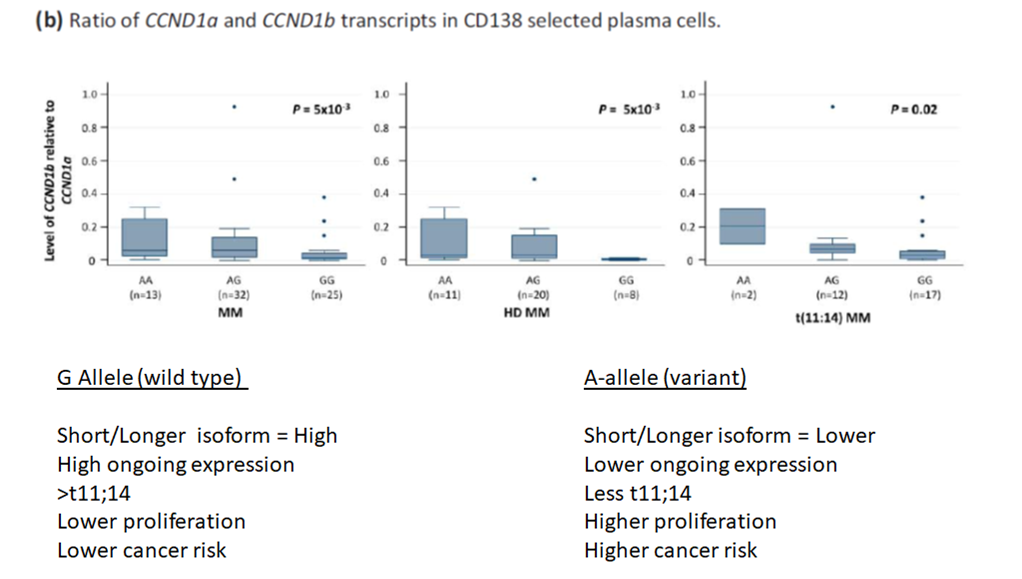
t11;14 translocations and splicing
We had showed a variant in CCND1 to be associated with increased risk of t11;14 translocation. As
the most significant variant is a described splice variant, we investigated the impact of the variant one splicing with the MM subgroups. Hyperdiplody and the t11;14
translocation lead to increased CCND1 expression, so in order to cleanly look at the association of the lead variant with aberrant splice it was essential to subgroup
experiments.
Programs and packages
Association testing - PLINK versions 1.7-1.9, IMPUTE, EIGENSTRAT and SNPTEST; Meta-analysis - PLINK, META, METAL; Data-visualisation - ggplot2, SNAP, matplotlib, seaborn; eQTL and meQTL - matrixQTL, FastQTL and PEER.
References
◦ Transcriptome-wide association study of multiple myeloma identifies candidate susceptibility genes. Went M, Kinnersley B, Sud A, Johnson DC, Weinhold N, Försti A, van Duin M, Orlando G, Mitchell JS, Kuiper R, Walker BA, Gregory WM, Hoffmann P, Jackson GH, Nöthen MM, da Silva Filho MI, Thomsen H, Broyl A, Davies FE, Thorsteinsdottir U, Hansson M, Kaiser M, Sonneveld P, Goldschmidt H, Stefansson K, Hemminki K, Nilsson B, Morgan GJ, Houlston RS. Hum Genomics. 2019 Aug 20;13(1):37. doi: 10.1186/s40246-019-0231-5. PMID: 31429796.
◦ Genetic Predisposition to Multiple Myeloma at 5q15 Is Mediated by an ELL2 Enhancer Polymorphism. Li N, Johnson DC, Weinhold N, Kimber S, Dobbins SE, Mitchell JS, Kinnersley B, Sud A, Law PJ, Orlando G, Scales M, Wardell CP, Försti A, Hoang PH, Went M, Holroyd A, Hariri F, Pastinen T, Meissner T, Goldschmidt H, Hemminki K, Morgan GJ, Kaiser M, Houlston RS. Cell Rep. 2017 Sep 12;20(11):2556-2564. PMID: 28903037
◦ Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Went M, Sud A, Försti A, Halvarsson BM, Weinhold N, Kimber S, van Duin M, Thorleifsson G, Holroyd A, Johnson DC, Li N, Orlando G, Law PJ, Ali M, Chen B, Mitchell JS, Gudbjartsson DF, Kuiper R, Stephens OW, Bertsch U, Broderick P, Campo C, Bandapalli OR, Einsele H, Gregory WA, Gullberg U, Hillengass J, Hoffmann P, Jackson GH, Jöckel KH, Johnsson E, Kristinsson SY, Mellqvist UH, Nahi H, Easton D, Pharoah P, Dunning A, Peto J, Canzian F, Swerdlow A, Eeles RA, Kote-Jarai Z, Muir K, Pashayan N, Nickel J, Nöthen MM, Rafnar T, Ross FM, da Silva Filho MI, Thomsen H, Turesson I, Vangsted A, Andersen NF, Waage A, Walker BA, Wihlborg AK, Broyl A, Davies FE, Thorsteinsdottir U, Langer C, Hansson M, Goldschmidt H, Kaiser M, Sonneveld P, Stefansson K, Morgan GJ, Hemminki K, Nilsson B, Houlston RS; PRACTICAL consortium. Nat Commun. 2018 Sep 13;9(1):3707. PMID: 30213928
◦ The CCND1 c.870G>A polymorphism is a risk factor for t(11;14)(q13;q32) multiple myeloma. Weinhold N*, Johnson DC*, Chubb D, Chen B, Försti A, Hosking FJ, Broderick P, Ma YP, Dobbins SE, Hose D, Walker BA, Davies FE, Kaiser MF, Li NL, Gregory WA, Jackson GH, Witzens-Harig M, Neben K, Hoffmann P, Nöthen MM, Mühleisen TW, Eisele L, Ross FM, Jauch A, Goldschmidt H, Houlston RS, Morgan GJ, Hemminki K. Nat Genet. 2013 May;45(5):522-525. *These authors contributed equally to this work. PMID: 23502783
◦ Multiple myeloma risk variant at 7p15.3 creates an IRF4-binding site and interferes with CDCA7L expression. Li N, Johnson DC, Weinhold N, Studd JB, Orlando G, Mirabella F, Mitchell JS, Meissner T, Kaiser M, Goldschmidt H, Hemminki K, Morgan GJ, Houlston RS. Nat Commun. PMID: 27882933.
◦ The 7p15.3 (rs4487645) association for multiple myeloma shows strong allele-specific regulation of the MYC-interacting gene CDCA7L in malignant plasma cells. Weinhold N*, Meissner T*, Johnson DC*, Seckinger A, Moreaux J, Försti A, Chen B, Nickel J, Chubb D, Rawstron AC, Doughty C, Dahir NB, Begum DB, Young K, Walker BA, Hoffmann P, Nöthen MM, Davies FE, Klein B, Goldschmidt H, Morgan GJ, Houlston RS, Hose D, Hemminki K. Haematologica. 2015 Mar;100(3):e110-3. *These authors contributed equally to this work. PMID: 25480495.